
Uniwersytet im. Adama Mickiewicza w Poznaniu
Wydział Filozoficzny
Zakład Logiki i Metodologii Nauk
email: tomasz.rzepinski@amu.edu.pl
Produkty lecznicze terapii zaawansowanych (ATMP) dają nadzieję na uzyskanie korzyści zdrowotnych w tych wszystkich sytuacjach, w których tradycyjne metody terapii zawodzą lub, z różnych przyczyn, nie mogą być stosowane. Zasadniczym celem niniejszego artykułu jest analiza pojęcia innowacyjności używanego w odniesieniu do biotechnologii wykorzystywanych w produkcji ATMP. Analiza tego pojęcia pozwoli wyodrębnić pięć głównych kontekstów znaczeniowych, które konstytuują jego sposób rozumienia: zmiana sposobu myślenia o dostępnym spektrum procedur medycznych, krótki czas rozpoznania technologii w modelach eksperymentalnych i teoretycznych, brak danych klinicznych dotyczących efektów stosowanych technologii w terapiach pacjentów, zwiększenie złożoności technologi oraz zwiększenie różnorodności stosowanych w tych technologiach substratów. Przeprowadzona analiza wskazuje na konieczność zachowania daleko idącej ostrożności w ocenie bezpieczeństwa terapii ATMP, w których wykorzystuje się innowacyjne technologie.
Słowa kluczowe: produkty lecznicze terapii zaawansowanych, biotechnologie inżynierii genetycznej, filozofia nauki, kategoria innowacyjności w medycynie
System opieki zdrowotnej musi być oparty na naukowych kryteriach oceny bezpieczeństwa i efektywności stosowanych oddziaływań terapeutycznych, diagnostycznych i prewencyjnych. Oczekiwać zatem należy, iż regulacje wprowadzane w tym systemie nadążają za rozwojem nowych technologii i odkryć w obszarze genetyki, immunologii i innych subdyscyplin biomedycznych1. Rozwój biotechnologii stworzył możliwość wykorzystania w medycynie zaawansowanych produktów inżynierii biomedycznej, jakimi są produkty terapii genowych, terapii komórkowych i inżynierii tkankowej. Dają one nadzieję na uzyskanie korzyści zdrowotnych w tych wszystkich sytuacjach, w których zawodzą, lub nie mogą, z różnych powodów, być stosowane tradycyjne metody terapii. Stosowanie produktów leczniczych zaawansowanych terapii medycznych (ATMP) stawia zupełnie nowe wyzwania zarówno przed pracownikami służby zdrowia, jak i instytucjami nadzorującymi i kontrolującymi wprowadzanie nowych terapii medycznych, które muszą podporządkować stosowane procedury do określonych wytycznych.
Pisząc o specyfice badań w obszarze ATMP należy przede wszystkim uświadomić sobie, że określenie „terapie innowacyjne” nie jest marketingowym ozdobnikiem, lecz odnosi się do rewolucyjnych zmian, które zapoczątkowane zostały pod koniec XX wieku i są kontynuowane do dzisiaj w obszarze badań biomedycznych. Zmiany te dają nie tylko nadzieję na poprawę długości i jakości życia pacjentów, ale stanowią również olbrzymie wyzwanie zarówno dla urzędów regulacyjnych, prowadzących nadzór nad rejestracją nowych produktów leczniczych, jak również dla zespołów badawczych dokonujących rozstrzygnięć metodologicznych, mających na celu określenie metod badawczych dostarczających najbardziej rzetelnych i wiarygodnych informacji na temat bezpieczeństwa i skuteczności produktów terapii zaawansowanych.
Zasadniczym celem niniejszego artykułu jest analiza pojęcia innowacyjności technologii, stosowanych w wytwarzaniu produktów leczniczych terapii zaawansowanych. W przeprowadzonej analizie tego pojęcia wyróżnione zostanie pięć głównych kontekstów znaczeniowych współtworzących sposób jego rozumienia. Po pierwsze, technologie innowacyjne wykorzystywane w terapii to technologie, które zmieniają w zasadniczy sposób myślenie o potencjalnych działaniach medycznych. Po drugie, ich innowacyjność polega na krótkim czasie ich rozpoznania w modelach eksperymentalnych i teoretycznych. Po trzecie, brakuje doświadczeń z ich wykorzystania w praktyce klinicznej. Po czwarte, innowacyjne technologie podlegają efektowi piętrzenia, czyli pewne technologie rozpoznawane jako innowacyjne mogą być doskonalone z wykorzystaniem innych technologii również posiadających innowacyjny charakter Po piąte, innowacyjność technologii polega na zróżnicowaniu substratów, które mogą być w nich zastosowane, a których efekty kliniczne nie są dobrze rozpoznane. Przeprowadzona analiza wskazuje na konieczność przyjęcia postawy daleko posuniętej ostrożności w ocenie bezpieczeństwa terapii ATMP, w których wykorzystuje się innowacyjne biotechnologie. Zasadne w tej sytuacji byłoby oczekiwanie, że innowacyjność terapii ATMP będąc pochodną stosowania w tych terapiach innowacyjnych technologii powinna być szczególnie dobrze kontrolowana w obszarze badań klinicznych. Problem polega jednak na tym, że jak pokazują dostępne do tej pory dane, oczekiwanie to nie jest spełnione2.
Termin „produkty lecznicze zaawansowanych terapii medycznych” odnosi się do heterogenicznej grupy produktów leczniczych, które różnią się zarówno procesem izolowania substratów, sposobami ich modyfikacji, jak i metodami stosowania, poczynając od pojedynczych iniekcji, a kończąc na działaniach o charakterze zabiegowym3. Termin ATMP obejmuje trzy główne rodzaje produktów: terapii genowych – Gene Therapy Medicinal Products (GTMP), somatycznych terapii komórkowych – somatic Cell Therapy Medicinal Products (sCTMP) oraz inżynierii tkankowej – Tissue-engineered Products (TEP).
Produkty terapii genowych (GTMP) to biologiczne produkty lecznicze:
Najogólniej, wszystkie terapie genowe mogą być podzielone na dwie kategorie. Pierwsza obejmuje terapie genowe linii zarodkowej, druga natomiast somatyczne terapie genowe. Zasadnicza różnica polega na tym, że w terapiach genowych linii zarodkowych wprowadzone zmiany genetyczne utrzymywane są w następnych pokoleniach, do czego nie dochodzi w somatycznych terapiach genowych. Jest to zgodne z dokumentem Rady Europy z 1997 roku, zgodnie z którym interwencja mająca na celu modyfikację ludzkiego genomu może być podjęta wyłącznie w celach profilaktycznych, diagnostycznych lub terapeutycznych i tylko wówczas, gdy jej celem nie jest wprowadzenie jakichkolwiek modyfikacji w genomie potomstwa5.
Produkty somatycznych terapii komórkowych (sCTMP) są biologicznymi produktami leczniczymi, które:
Produkty ATMP mogą różnić się źródłem pozyskania materiału. W transplantacjach autologicznych wykorzystane zostają odpowiednio modyfikowane komórki własne pacjenta, który pełni wówczas podwójną rolę: dawcy i biorcy. Przykładem jest terapia CAR-T (patrz paragraf 6). Limfocyty T izolowane z krwi pacjenta chorego na raka są modyfikowane genetycznie w celu uzyskania ekspresji receptorów wiążących antygeny nowotworowe. Następnie podawane są z powrotem temu samemu pacjentowi. Komórki T atakują wówczas komórki nowotworowe wykazujące ekspresję danego antygenu. Z kolei w transplantacjach allogenicznych CTMP zostaje pobrane od zdrowej osoby pełniącej funkcję dawcy7.
Produkty inżynierii tkankowej (TEP) to produkty spełniające następujące warunki:
Produkt inżynierii tkankowej może zawierać komórki lub tkanki pochodzenia ludzkiego lub zwierzęcego, lub pochodzące z obu tych źródeł. Komórki lub tkanki mogą być żywotne lub nieżywotne. Może on także zawierać dodatkowe substancje, takie jak produkty komórkowe, biomolekuły, biomateriały, substancje chemiczne8.
Zarówno sCTMP jak i TEP są produktami inżynierii, które muszą spełniać jeden z dwóch warunków:
Produkt leczniczy terapii zaawansowanej zawierający komórki lub tkanki zarówno autologiczne (pochodzące od samego pacjenta), jak i allogeniczne (pochodzące od innego człowieka) uważa się za przeznaczony do użytku allogenicznego10.
Dodatkowo ATMP może również obejmować łączne zastosowanie co najmniej dwóch z wyżej wymienionych terapii, są to tzw. łączone ATMP (combined – cATMP)11. Przykładem mogą być projekty terapii łączących inżynierię genową i terapię z wykorzystaniem komórek macierzystych w celu leczenia uszkodzeń płodu12. Podkreślić jednak należy, że cATMP jest terminem „technicznym”, stosowanym w obszarze prowadzonych badań naukowych, natomiast nie ma uzasadnienia legislacyjnego, ponieważ Rozporządzenie 1394/2007 Parlamentu Europejskiego z dnia 13 listopada 2007 r wyraźnie określa sposób klasyfikowania różnych terapii cATMP do jednej z głównych grup. W szczególności:
Należy wyraźnie zaznaczyć, że ustalenie, jaki typ produktu reprezentuje dany ATMP należy do kompetencji Komitetu ds. Terapii Zaawansowanych (Committee for Advanced Therapies, CAT) i nie zawsze jest oczywiste, w szczególności w odniesieniu do rozróżnienia pomiędzy sCTMP i GTMP. Przykładem ilustrującym tę kwestię może być porównanie dwóch produktów leczniczych ATMP – StrimvelisuR i ZalmoxisuR. W obu przypadkach wprowadzono do komórek krwi wektor retrowirusa z odpowiednio zmodyfikowanym materiałem genetycznym. Pierwszy produkt jest wykorzystywany w terapii ciężkiego złożonego braku odporności (ADA-SCID). Modyfikacje genetyczne miały na celu wprowadzenie funkcjonalnych zmian w pewnym genie odpowiedzialnym za produkcję enzymu ADA u pacjentów z ADA-SCID. Dlatego StrimvelisR jest klasyfikowany jako GTMP. Z kolei ZalmoxisR jest stosowany jako leczenie wspomagające u pacjentów po przeszczepie. W efekcie przeszczepu dochodzi u niektórych pacjentów do zdarzeń niepożądanych związanych z nadmierną indukcją układu immunologicznego (jest to tzw. choroba przeszczep przeciwko gospodarzowi, GvHD). Wprowadzona w tym produkcie modyfikacja genetyczna miała na celu wyłącznie wprowadzenie genu samobójczego do komórek, którego aktywacja w przypadku wystąpienia GvHD za pomocą odpowiedniego leku wirostatycznego doprowadziłaby do śmierci komórek użytych w ZalmoxisieR. Lek ten jest klasyfikowany nie jako GTMP, tylko jako sCTMP, ponieważ zmiany genetyczne nie mają na celu realizacji zasadniczego celu terapii, lecz pozostawiają „okno możliwości” do podjęcia działania w przypadku wystąpienia zdarzenia niepożądanego terapii13.
W Europie w 2003 roku przyjęto dyrektywę, na podstawie której ATMP zostały zdefiniowane jako „oparte na procesach produkcyjnych ukierunkowanych na różne transfery genów kodujących syntezę biomolekuł i / lub biologicznie zaawansowanych modyfikacjach terapeutycznych komórek jako substancji czynnych, lub części substancji czynnych”14. W konsekwencji TEP były nieobjęte tym rozporządzeniem. Zasadniczy przełom w ustawodawstwie unijnym dotyczącym ATMP nastąpił w 2007 roku, gdy dyrektywa definiująca terapie zaawansowane została odpowiednio zmieniona na podstawie rozporządzenia EC No. 1394/2007, tak aby terapie ATMP obejmowały również TEP15.
Na mocy Rozporządzenia 1394/2007 wyodrębniono w ramach Europejskiej Agencji Leków (European Medicines Agency, EMA) Komitet ds. Terapii Zaawansowanych (Committee for Advanced Therapies, CAT). Wyodrębnienia tego Komitetu dokonano stwierdzając, że ocena produktów leczniczych terapii zaawansowanej często wymaga bardzo konkretnych kompetencji, wykraczających poza tradycyjną farmację i obejmujących obszary z pogranicza innych sektorów, takich jak biotechnologia i wyroby medyczne16.
Kolejne rozporządzenia mają na celu nie tyle doprecyzowanie definicji ATMP, lecz raczej przede wszystkim ustalenie wymagań i procedur dopuszczenia do obrotu produktów terapii zaawansowanych ze szczególnym uwzględnieniem sposobów oceny skuteczności, bezpieczeństwa i zarządzania ryzykiem w obszarze tych działań medycznych z ich wykorzystaniem. Jednostką odpowiedzialną za naukową ocenę wniosków o pozwolenie na dopuszczenie produktów terapii zaawansowanych do obrotu (marketing authorisation – MA) jest wspomniany wcześniej Komitet ds. Terapii Zaawansowanych (CAT).
Głównym zadaniem CAT jest opiniowanie wniosków ATMP złożonych do EMA zanim Komitet ds. Produktów Leczniczych Stosowanych u Ludzi (Committee for Medicinal Products for Human Use, CHMP) wyda opinię w sprawie dopuszczenia leku do obrotu. W tym zakresie zasadniczym obowiązkiem CAT jest rozstrzygnięcie, czy dany środek spełnia definicję i powinien uzyskać status ATMP. Warto podkreślić, że pomimo stosowania definicji precyzujących ATMP nie zawsze takie rozpoznanie jest oczywiste, o czym świadczy fakt, że dużo produktów nie jest przez CAT uznawanych za ATMP17.
Do zadań CAT należy również udzielanie porad w kwestii oceny bezpieczeństwa, skuteczności i zarządzania ryzykiem w odniesieniu do wytwarzania, przechowywania, obrotu i stosowania ATMP; formułowanie ekspertyz oraz wspieranie rozwoju nowych ATMP18.
Regulacje UE ustalają również bardzo wyraźnie, że produkty lecznicze terapii zaawansowanych powinny podlegać tym samym ogólnym regulacjom, jakim podlegają inne typy produktów medycznych, w szczególności przestrzegane muszą być ogólne normy jakości i bezpiecznego oddawania, pobierania, testowania, przetwarzania, konserwowania, przechowywania i dystrybucji komórek i tkanek ludzkich. Wskazuje się jednak również, że mogą istnieć szczegółowe wymagania techniczne, w szczególności w odniesieniu do rodzaju i ilości jakościowych danych przedklinicznych i klinicznych niezbędnych do wykazania jakości, bezpieczeństwa i skuteczności produktu19.
Z perspektywy celów realizowanych w niniejszym artykule ważne jest również zwrócenie uwagi na wytyczne dotyczące badań klinicznych ATMP, które zgodnie z przyjętym rozporządzeniem powinny być prowadzone w sposób uwzględniający ogólne zasady dobrej praktyki klinicznej w prowadzeniu badań klinicznych produktów leczniczych przeznaczonych do stosowania u człowieka. W szczególności podkreśla się, że to właśnie kontrola bezpieczeństwa i skuteczności ATMP jest kluczowym aspektem regulacji dotyczących tych produktów leczniczych20.
W 1990 r. amerykańska Agencja ds. Żywności i Leków (Food and Drug Administration, FDA) zatwierdziła pierwsze badanie kliniczne terapii genowej. Dwóm pacjentom pediatrycznym z ciężkim wrodzonym brakiem odporności w wyniku niedoboru deaminazy adenozynowej (ADA-SCID) podano autologiczne, zmodyfikowane ex vivo (poza ustrojem) krwinki białe. ADA-SCID jest chorobą monogenową, czyli spowodowaną mutacją w pojedynczym genie. Terapia okazała się wprawdzie bezpieczna, ale niestety mało skuteczna. U jednej z pacjentek stwierdzono poprawę, u drugiej poprawy nie zaobserwowano21. W 1999 r pacjentowi z niedoborem transkarbamylazy ornityny (OTC) podano andenowirus przenoszący brakujący u pacjenta gen. Pacjent zmarł w efekcie nadczynności systemu immunologicznego, prowadzącej do nieodwracalnych uszkodzeń narządów wewnętrznych22. Kolejna próba terapii genowej doprowadziła do rozwoju wtórnej białaczki u kilkorga pacjentów pediatrycznych23. Pierwszym krajem, który zatwierdził stosowanie terapii genowej były Chiny. Zaaprobowanym lekiem był GendicineR stosowany u pacjentów z pewnymi rodzajami raków płaskonabłonkowych spowodowanymi mutacjami w pojedynczym genie. Terapia polegała na wprowadzeniu odpowiednio zmodyfikowanego adenowirusa, który stymulował śmierć komórek rakowych.
W 2015 roku, czyli 8 lat po przyjęciu wspomnianego wcześniej rozporządzenia EC No. 1394/2007, zaledwie 5 terapii typu ATMP dopuszczono do obrotu handlowego na terenie Unii Europejskiej. Była to jedna somatyczna terapia komórkowa (ProvengeR) raka gruczołu krokowego dopuszczona na rynek w 2013 roku; jedna terapia genowa (GlyberaR) dopuszczona w 2012 roku w przypadkach uwarunkowanego genetycznie niedoboru lipazy lipoproteinowej oraz trzy autologiczne terapie inżynierii tkankowej: ChondroCelectR w 2009, MACIR w 2013 (klasyfikowany również jako cATMP24) oraz HoloclarR w 2015 roku, będący pierwszą zarejestrowaną w UE terapią komórkami macierzystymi25. Podkreślić należy, że zarówno Glybera jak i Holoclar zostały zakwalifikowane jako leki sieroce. Holoclar uzyskał warunkowe dopuszczenie do obrotu26.
Ta bardzo skrócona historia pierwszych prób związanych z terapiami, które współcześnie kwalifikowane są jako terapie ATMP wskazuje na dwie ważne kwestie. Po pierwsze, jak niewiele czasu w gruncie rzeczy minęło od tych prób. Po drugie, jak bardzo mało efektywne były te pierwsze terapie spełniające charakterystykę ATMP.
W pierwszych latach po wprowadzeniu regulacji prawnych UE wyraźnie zwiększyła się dynamika rozwoju nowych ATMP. Widać to na podstawie wyników przeglądu systematycznego, jakiego w 2016 roku dokonali E. Hanna i współpracownicy. W oparciu o przyjęte w 2007 roku regulacje charakteryzujące terapie ATMP w Unii Europejskiej, autorzy zidentyfikowali 939 badań klinicznych prowadzonych na całym świecie w obszarze terapii ATMP w latach 1999-2015. Z tego 504, czyli ponad 53% to badania kliniczne, których przedmiotem były produkty somatycznych terapii komórkowych, 210 (22,4%) to badania kliniczne terapii genowych, 214 (22,8%) badań dotyczyło terapii w zakresie inżynierii tkankowej, natomiast zaledwie 11 (1,2%) dotyczyło terapii łączonych cATMP. Należy zwrócić uwagę, że identyfikacja tych badań klinicznych produktów ATMP, które były przeprowadzone w latach 1999-2007, miała charakter retrospektywny. Dla potrzeb analizy autorzy przyjęli ustalenia wprowadzone w 2007 roku stanowiące podstawę rozstrzygnięć dokonywanych przez CAT27. Retrospektywna analiza wykazała, że przyjęte w 2007 roku kryteria oceny pozwalają zakwalifikować 112 badań klinicznych przeprowadzonych w latach 1999–2007 jako badania produktów ATMP (patrz schemat 1 poniżej). Nie daje to oczywiście gwarancji, że wszystkie te terapie byłyby zatwierdzone przez CAT jako ATMP, ale wskazuje wyraźnie na fakt, że w pewnym okresie rozwoju nowych technologii pojawiła się w środowiskach medycznych potrzeba wyraźnego legislacyjnego odróżnienia działań medycznych typu ATMP od standardowych rodzajów terapii.
Dynamikę rozwoju badań klinicznych ATMP reprezentuje poniższy schemat.
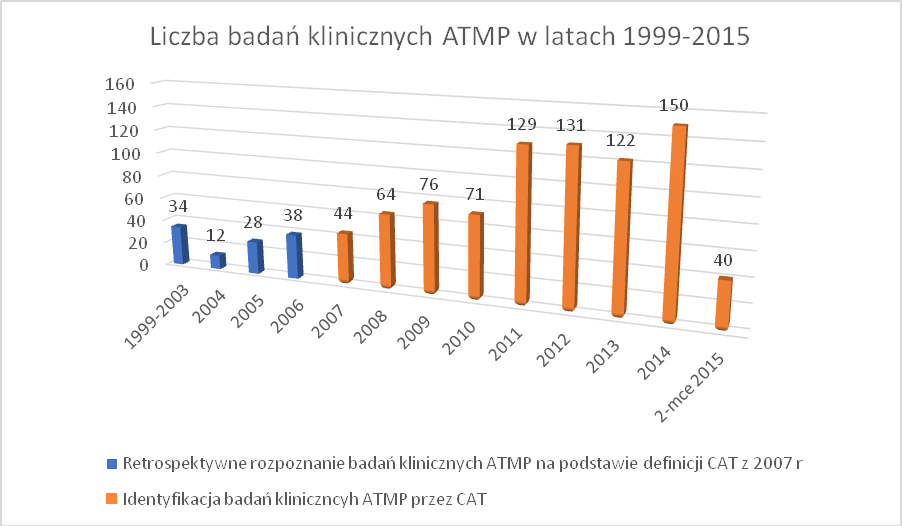
Przeprowadzona przez Hannę i współpracowników analiza pozwoliła stwierdzić, że dwoma głównymi obszarami przeprowadzanych badań klinicznych w obszarze terapii ATMP były: onkologia 24,8% (w tym nowotwory hematologiczne (30,1%), skóry (10,1%), raka gruczołu krokowego (9,9%), mózgu (8,2%), gastroenterologiczne (7,7%)) oraz kardiologia 19,4% (z czego niewydolność serca i kardiomiopatie (31,3%), niedokrwienie kończyn (24,2%), zawał serca (23,6%)). Pozostałe badania kliniczne obejmowały między innymi choroby układu immunologicznego (11,5%), choroby układu mięśniowo-szkieletowego (10,5%), choroby neurologiczne (9,1%), cukrzyce (5,2%). Patrz schemat 2.
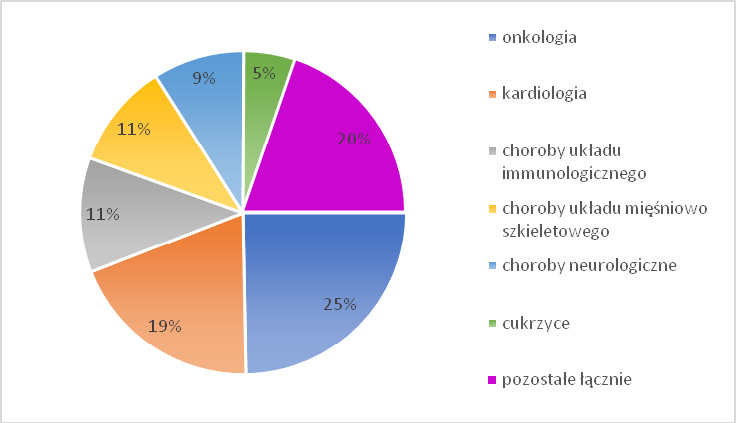
Zauważmy, że zgodnie z powyższym schematem największa liczba badań klinicznych ATMP dotyczyła onkologii. W tym obszarze były to przede wszystkim GTMP, których kontrowersyjność związana jest z zastosowaniem innowacyjnych technologii inżynierii genetycznej. Przyjrzyjmy się zatem bliżej tej kwestii.
Wśród produktów ATMP najwięcej kontrowersji i problemów w obszarze prowadzonych badań budzą produkty terapii genowych. Powodem są nie tylko obawy natury etycznej, ale również, a być może przede wszystkim, innowacyjność stosowanych technologii, co w wymierny sposób przekłada się zarówno na problemy oceny bezpieczeństwa i skuteczności terapii zaprojektowanych z wykorzystaniem tych technologii, jak i na problemy legislacyjne. Do najbardziej innowacyjnych technologii GTMP należą technologie wykorzystujące: wektory pełniące funkcję nośników materiału genetycznego, komórki CAR-T, interferujące RNA oraz elementy genomu bakterii CRISPR/Cas. W dalszej części artykułu stosowane będą określenia „technologia wektorów”, „technologia CAR-T”, „technologia RNAi” oraz „technologia „CRISPR/Cas”. Wyrażenia te będą odnosiły się do technologii, w których wykorzystano wskazane elementy. Przyjrzyjmy się im zatem bliżej.
Najogólniej „wektorami” określane są nośniki materiału genetycznego (najczęściej wirusy), które mogą zostać użyte w terapii poprzez wprowadzenie ich do komórki w celu modyfikacji jej materiału genetycznego. Po wprowadzeniu następuje zapoczątkowanie procesów, które prowadzą do wytwarzania białka terapeutycznego i w efekcie umożliwiają leczenie pacjenta.
Technologia CAR-T wykorzystywana jest w celu modyfikacji komórek układu odpornościowego, limfocytów T, tak aby wytwarzały one receptor antygenowy, zwany chimerycznym receptorem antygenowym (CAR), dzięki któremu limfocyty mają zdolność rozpoznawania komórek nowotworowych. Zmodyfikowane limfocyty podawane są następnie pacjentowi. Technologia CAR-T pozwala zatem w odpowiedni sposób „wyuczyć” i zaprogramować system odpornościowy pacjenta do działań mających na celu wyeliminowanie komórek nowotworowych, które wcześniej nie były przez elementy tego systemu rozpoznawane. Terapia wykorzystująca technologię CAR-T okazała się niezwykle skuteczna w leczeniu białaczki limfoblastycznej i szpiczaka mnogiego30.
Kolejną ważną technologią stosowaną w terapiach genowych jest metoda hamowania ekspresji genów z wykorzystaniem niskocząsteczkowego interferującego RNAi. W procesie ekspresji genów informacja genetyczna zostaje przekształcona na określone cząsteczkowe produkty, jak białka i RNA. W przypadku chorób genetycznych celem terapii może być zahamowanie ekspresji wadliwych genów. Technologia RNAi umożliwia takie właśnie wyłączenie / wyciszenie genu odpowiedzialnego za produkcję niewłaściwego białka, np. białka o błędnej strukturze31.
Ważną technologią stosowaną w terapiach genowych jest również metoda CRISPR/Cas32. Technologia ta reprezentuje zupełnie odmienną od wcześniejszych strategię modyfikowania genów. Pozwala bowiem na korygowanie aberracji genetycznej bezpośrednio in situ. CRISPR i Cas są elementami genomu bakterii. Wykorzystanie połączenia obu tych elementów pozwoliło stworzyć niezwykle efektywne narzędzie edycji genów33.
Działania medyczne, w których wykorzystywane są ATMP określane są bardzo często mianem terapii innowacyjnych. W 2014 roku powołana została w ramach EMA jednostka Innovation Task Force (ITF), której przedmiotem analiz są innowacyjne terapie i technologie. ITF posiada charakter interdyscyplinarny, skupiając badaczy z różnych dyscyplin, zarówno biologów, genetyków, immunologów, klinicystów, jak i prawników. Do jej głównych zadań należą:
Mogłoby się wydawać, że mówiąc o terapiach innowacyjnych mamy po prostu na myśli terapie nowe, w których stosuje się substancje aktywne, które wcześniej nie były wykorzystywane w praktyce medycznej. Można wówczas orzec innowacyjność terapii w odniesieniu do każdej „klasycznej” terapii farmakologicznej, w której zastosowana jest nowo zsyntetyzowana cząsteczka, nigdy wcześniej nie zarejestrowana i której zadaniem jest hamowanie lub aktywacja pewnego szlaku biochemicznego poprzez odpowiednie oddziaływanie na receptory komórkowe.
Nie ulega jednak wątpliwości, że mówiąc o „innowacyjności” produktów terapii zaawansowanych, w szczególności GTMP, mamy na myśli coś więcej. Innowacyjność tych terapii jest bowiem w istocie pochodną innowacyjności biotechnologii, które są wykorzystywane przy wytwarzaniu tych terapii. Z perspektywy filozofa nauki interesujące jest zatem ustalenie, na czym polega „innowacyjność technologii”. Co mają na myśli badacze posługujący się tym pojęciem? W jakiej relacji nowe technologie stosowane w nauce, w tym również w medycynie, muszą pozostawać do obowiązującej wiedzy naukowej, abyśmy skłonni byli określić je mianem innowacyjnych? Czy innowacyjność oznacza po prostu słabe „rozpoznanie” tych technologii, niewystarczające ich osadzenie w obszarze teorii nauk podstawowych? Jeżeli tak, to jakie jest uzasadnienie dla wykorzystywania tych technologii w praktyce medycznej? Z perspektywy analiz epistemologicznych rysuje się tu wyraźny rozdźwięk pomiędzy, z jednej strony, uzasadnieniem, które pozwala nam wykorzystywać te technologie w praktyce medycznej, a postawą ostrożności epistemicznej, nakazującej nam posługiwanie się w odniesieniu do tych terapii określeniem innowacyjne.
Analizując pojęcie „innowacyjności” stosowane w odniesieniu do technologii biomedycznych wykorzystywanych w terapiach GTMP można wskazać na pięć podstawowych kontekstów znaczeniowych konstytuujących sposób rozumienia tego pojęcia. Są nimi odpowiednio:
W odniesieniu do pierwszego punktu należy zauważyć, że dla filozofa nauki innowacyjność jest związana z radykalną zmianą sposobu myślenia o sposobach rozwiązywania pewnych problemów lub stosowaniu pewnych narzędzi. Przejście od ciągnionych sań do konstrukcji wózka z kołami, czy od pomiaru czasu z wykorzystaniem świec, klepsydr czy zegarów słonecznych do wprowadzenia wychwytu Grahama umożliwiającego precyzyjne uwalnianie energii potencjalnej ciężarków lub sprężyn mechanizmów zegarowych, to jedne z najbardziej oczywistych przykładów ilustrujących taki właśnie sposób rozumienia kategorii innowacyjności. Problem epistemologiczny cały czas jednak pozostaje. Nie określilibyśmy współcześnie wózka z kołami czy mechanizmu zegarowego mianem innowacyjnych. Podobnie, można wnosić, że pewne technologie biomedyczne, które uznawane są obecnie za innowacyjny fundament terapii medycznych, w przyszłości utracą swój innowacyjny status, co wyraźnie wskazuje na fakt, że innowacyjność związana jest z pewną dozą niepewności co do konsekwencji ich stosowania.
Drugi z wymienionych kontekstów związany jest z krótkim okresem rozpoznania nowych technologii. Zauważmy, że leżąca u podstaw klasycznej farmakoterapii idea blokowania lub aktywowania szlaków sygnałowych jest rozwijana od ponad stu lat. Tymczasem idea, w której działania medyczne mają być oparte na odpowiednio przeprowadzonych modyfikacjach materiału genetycznego jest w praktyce rozwijana w badaniach naukowych raptem od kilku dekad.
Przykładowo, wykorzystanie wirusów jako nośników materiału genetycznego to przełom lat 80. i 90. XX wieku35. Opracowanie technologii CAR-T to pierwsza dekada XXI wieku. Odkrycie interferujących RNA zostało dokonane dopiero w 1998 roku.36 Technologia CRISPR/Cas opracowana została również na przełomie XX i XXI wieku37, ale pierwsze badania na liniach komórkowych ssaków z wykorzystaniem tej technologii to dopiero 2013 rok.38
Krótki czas rozpoznania innowacyjnych technologii inżynierii genetycznej wpływa w oczywisty sposób na niewielkie doświadczenia z ich zastosowania w praktyce medycznej. Pierwsza terapia, w której wykorzystano wirus jako nośnik materiału genetycznego wprowadzonego do ustroju pacjenta, została przeprowadzona w 1990 roku (terapia dwóch pacjentek, efektem było uzyskanie umiarkowanej korzyści terapeutycznej). Kolejna dopiero w 1999 roku (terapia pojedynczego pacjenta, doprowadziła do jego śmierci – przypadek niedoboru OTC wspomniany w paragrafie 4 powyżej). W 2017 roku zatwierdzona została przez FDA terapia ATMP białaczki limfoblastycznej (lek Kymriah), wykorzystująca technologię CAR-T (w Polsce refundowana od 2021). W 2018 roku zatwierdzone zostały przez FDA dwa leki oparte na technologii RNAi: OnpattroR i TegsediR dla potrzeb leczenia dziedzicznej amyloidozy transtyretynowej. Z kolei dopiero w 2019 roku FDA zatwierdziła terapię ATMP opartą na technologii CRISPR/Cas w leczeniu anemii sierpowatej, czyli choroby genetycznej prowadzącej do powstawania nieprawidłowo zbudowanych komórek hemoglobiny39. Również w 2019 roku rozpoczęto badania kliniczne nad użyciem technologii CRISPR/Cas w terapii ślepoty Lebera40. Wyraźnie zatem widoczne jest, że rozpoznawanie efektów klinicznych stosowania innowacyjnych biotechnologii w leczeniu chorych następowało w stosunkowo krótkim czasie.
Innowacyjność technologii stosowanych w inżynierii genetycznej związana jest też z efektem zwiększania złożoności technologii. Stosowane od niedawna innowacyjne biotechnologie inżynierii genetycznej są usprawniane z wykorzystaniem kolejnych, których czas rozpoznania w badaniach podstawowych jest jeszcze krótszy. Przykładem może być technologia CRISPR/Cas, która umożliwia zwiększenie funkcjonalności technologii CAR-T41. Niemniej jednak doskonale wiadomo, że każde zwiększenie złożoności mechanizmów wytwarzania ATMP zwiększa ryzyko, że któryś z elementów tych mechanizmów zawiedzie i efekty kliniczne będą odmienne od oczekiwanych. Wraz ze zwiększeniem złożoności mechanizmów zmniejsza się prawdopodobieństwo trafnego przewidywania zdarzeń klinicznych.
Ostatnią ważną cechą, którą należy wymienić, gdy rozważamy innowacyjność technologii zastosowanych w ATMP, jest zróżnicowanie stosowanych w tych technologiach substratów. Przykładem może być wspomniana wcześniej technologia, w której funkcje wektorów pełnią najczęściej, choć nie zawsze, różnego typu wirusy: retrowirusy, adenowirusy, lentiwirusy, wirusy AAV, czyli tzw. wirusy zależne od adenowirusów42. Wykorzystanie różnych wirusów jako wektorów danej terapii genowej przynosi określone korzyści, ale niesie ze sobą jednocześnie ryzyko terapeutyczne i podlega pewnym ograniczeniom z uwagi na realizowane cele terapii. Wybór rodzaju wektora jako nośnika informacji genetycznej musi być zatem podyktowany dogłębną oceną korzyści i ryzyka związanego z jego zastosowaniem. Przykładowo retrowirusy nie wykazują zwiększonego działania immunogennego, czyli nadmiernego pobudzenia systemu immunologicznego pacjenta, które ma miejsce w przypadku wirusów AAV i adenowirusów. W przypadku tych dwóch ostatnich może bowiem nastąpić nadmierna odpowiedź immunologiczna u pacjentów, którzy z racji na przebyte wcześniej infekcje mieli już kontakt z tymi wirusami. Ocenia się, że ponad 80% populacji posiada przeciwciała przeciwko adenowirusom. W skrajnych przypadkach terapia wykorzystująca adenowirusy jako wektory może okazać się tragiczna w skutkach, jak miało to miejsce w przypadku drugiej historycznie terapii genowej zastosowanej u pacjenta z zespołem OTC w 1999 roku (patrz paragraf 4).
Innym przykładem zróżnicowania stosowanych substratów jest technologia CRISPR/Cas. Jednym z głównych problemów tej technologii jest występowanie dużej immunotoksyczności spowodowanej faktem, że znaczna część populacji miała kontakt z różnymi typami bakterii Streptococcus lub z wirusami AAV wykorzystywanymi w tej technologii jako wektory. Dlatego też technologia CRISPR/Cas podlega nieustannemu rozwojowi. Obecnie prowadzone są badania nad 30 wariantami elementu Cas9, które są uzyskiwane z trzech różnych rodzajów bakterii Streptococcus43. Warianty te różnią się pewnymi cechami, o których sądzi się, że mogą wpływać na efekty kliniczne zastosowanej technologii w wytwarzaniu ATMP. Przykładów zróżnicowania substratów wykorzystywanych w ATMP dostarczają również somatyczne terapie komórkowe, wśród których wykorzystuje się macierzyste komórki embrionalne, indukowane pluripotencjalne komórki macierzyste, komórki mezenchymalne, rąbkowe komórki macierzyste stosowane w okulistyce, czy nerwowe komórki macierzyste, w których pokłada się duże nadzieje w związku z terapiami chorób neurologicznych44.
Innowacyjność technologii stosowanych w inżynierii genetycznej jest konstytuowana w każdym z wymienionych w poprzednim paragrafie aspektów. Za sprawą tych technologii dokonała się zasadnicza zmiana sposobu myślenia o działaniach medycznych, które można podejmować dla dobra pacjenta. Technologie te tworzą zupełnie odmienną od dominującej do tej pory perspektywę postrzegania sprawczości działań medycznych. Po wielu dekadach rozwoju klasycznej farmakologii, której celem było wprowadzanie do organizmu naturalnych lub zsyntetyzowanych cząsteczek biologicznie aktywnych w celu wywołania określonego efektu terapeutycznego, profilaktycznego, lub diagnostycznego, realizuje się idea oddziaływań prozdrowotnych poprzez wprowadzanie do ustroju produktów stanowiących rezultat manipulacji materiałem genetycznym, lub produktów dokonujących takich modyfikacji bezpośrednio w ustroju pacjenta. Zmianę tego sposobu myślenia można najdobitniej porównać do zmian, jakie nastąpiły w II połowie XIX wieku wraz z przesunięciem uwagi z obszaru terapii na obszar oddziaływań profilaktycznych tj. rozwoju badań L. Pasteura nad szczepieniami. W obu przypadkach mamy do czynienia z rewolucyjną zmianą w sposobie myślenia o celach oddziaływań medycznych. Cele, które nie były osiągalne z udziałem klasycznych terapii farmakologicznych mogą być współcześnie realizowane dzięki terapiom opartym na technologiach stosowanych w ATMP. Opisany rozwój głównych technologii stosowanych w GTMP ilustruje poniższy schemat45.
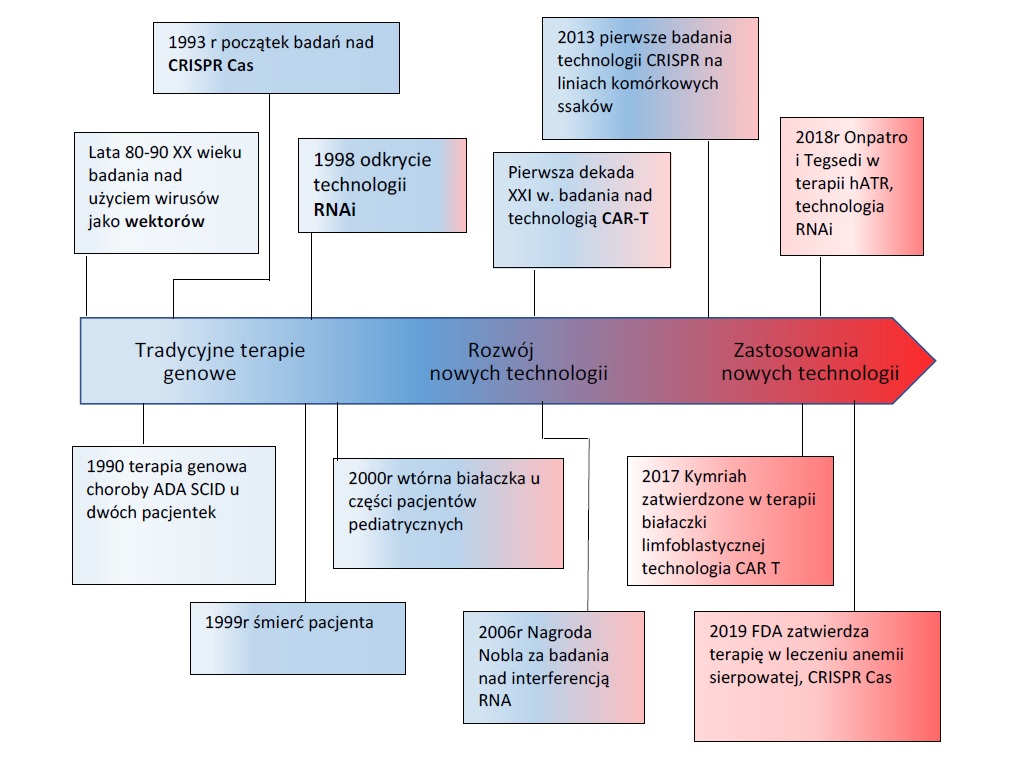
Na aspekt innowacyjności technologii stosowanych we współczesnej medycynie zwraca uwagę Buckland pisząc:
obecnie mamy do czynienia z technologiczną rewolucją w medycynie. Odkrycie ludzkiego genomu i rozwój technologii sekwencjonowania genów doprowadziły do zrozumienia przyczyn rozwoju wielu chorób dziedzicznych. Mamy możliwość zaobserwowania niezwykłych postępów technologicznych umożliwiających podejmowanie oddziaływań genetycznych46.
Oczywiste pytanie, które należy w tym kontekście postawić dotyczy tego, jak wskazane aspekty innowacyjności technologii stosowanych w GTMP wpływają na ocenę przede wszystkim bezpieczeństwa oraz, w drugim rzędzie, skuteczności terapii.
Problemem innowacyjnych technologii nie jest to, że pojawiają się nowe zagrożenia, z którymi wcześniej nie mieliśmy do czynienia, a które chcemy, choćby z dużą niepewnością, brać pod uwagę w projekcie terapii. Problemem, zarówno z perspektywy klinicysty, jak i z perspektywy epistemologicznej jest to, czego posiadana przez nas wiedza nie obejmuje w ogóle, czyli te konsekwencje stosowania innowacyjnych technologii, o których nie posiadamy najmniejszych nawet wyobrażeń, ponieważ nasza wiedza teoretyczna o nowych technologiach nie jest dostatecznie dobrze ugruntowana, ani w systemach teoretycznych tworzonych na podstawie badań naukowych, ani w praktyce zastosowań klinicznych tych technologii. W ocenie obu tych kontekstów posługujemy się pojęciem ryzyka, którego wartość określana jest z wykorzystaniem pojęcia prawdopodobieństwa. Celem jest ustalenie wartości, lub przynajmniej przybliżone oszacowanie akceptowanego stopnia ryzyka wystąpienia danego zdarzenia klinicznego. Pamiętać jednak należy, że w obu tych kontekstach znaczenia tego pojęcia są różne, posiadając ugruntowanie w odmiennych w istocie interpretacjach prawdopodobieństwa.
Przede wszystkim należy zauważyć, że pojęcie akceptowalnego stopnia ryzyka odnosi się do zdarzeń, o których mamy jakiekolwiek wyobrażenie. Termin ten nie ma natomiast żadnego zastosowania wobec zdarzeń, z którymi nigdy wcześniej nie mieliśmy do czynienia. Problemem nie jest zatem to, że stosowanie adenowirusów jako wektorów może być przyczyną nadmiernej aktywacji układu immunologicznego prowadząc do uszkodzeń wielonarządowych, lub nawet śmierci. To już wiadomo, jakkolwiek warto podkreślić, że nawet w tym przypadku świadomość zagrożeń nie uchroniła przed katastrofalną konsekwencją zastosowania jednej z pierwszych terapii genetycznych w 1999 roku. Przypadek ten jest przykładem błędnego określenia wartości akceptowalnego stopnia ryzyka. Błędnego, ponieważ były podstawy (wiedza z modeli zwierzęcych) do wnioskowania o wystąpieniu zjawiska nadmiernej odpowiedzi immunologicznej47. Problemem znacznie poważniejszym niż poprawna ocena ryzyka akceptowalnego są jednak te zdarzenia niekorzystne medycznie, o których wystąpieniu nie mamy żadnych wyobrażeń, bądź to z uwagi na zbyt dużą złożoność zastosowanych technologii, niewystarczającą wiedzę o efektach stosowania różnych substratów / elementów, czy po prostu za względu na krótki czas rozpoznania technologii w praktyce medycznej.
Różnicę pomiędzy obydwoma kontekstami można zilustrować odnosząc się do częstościowej i personalistycznej interpretacji prawdopodobieństwa. Jak zwracał uwagę twórca tej pierwszej i główny jej propagator, von Mises, zasadniczą wadą tej interpretacji jest to, że nie pozwala oszacować prawdopodobieństwa zdarzeń, z którymi nigdy nie mieliśmy do czynienia. Nie można z jej wykorzystaniem ocenić prawdopodobieństwa wybuchu nowej wojny ani istnienia życia w kosmosie. Można ją wykorzystywać wyłącznie w celu oceny prawdopodobieństwa wystąpienia zdarzeń, które miały już wcześniej miejsce48. Interpretacja ta jest zatem podstawą dla definiowania bez mała wszystkich współczynników stosowanych w epidemiologii. Pojęcie akceptowalnej wartości ryzyka zdarzenia klinicznie niekorzystnego dotyczy również tego rodzaju zdarzeń, których prawdopodobieństwo może być określone w sposób częstościowy, lub da się przynajmniej dokonać wiarygodnych estymacji tego prawdopodobieństwa na podstawie wiedzy z modeli zwierzęcych.
Tymczasem ocena ryzyka zdarzeń niekorzystnych, o których nie posiadamy żadnej wiedzy, a które nigdy nie wystąpiły, wymagałaby skrajnie subiektywistycznej interpretacji prawdopodobieństwa. Zauważmy, że większość tzw. „katastrof medycznych” nie była efektem zlekceważenia problemu lub oszustwa, lecz stanowiła konsekwencję braku wiedzy, braku jakichkolwiek przesłanek co do możliwości wystąpienia niekorzystnych efektów podejmowanych działań medycznych. Wystarczy przywołać historię tragedii zastosowania leków antyarytmicznych u osób po zawale serca, lub nieprzewidywalne efekty zaobserwowane w badaniu HERS II zastępczej terapii hormonalnej. Podobnie rzecz ma się w odniesieniu do innowacyjnych technologii stosowanych w medycynie. Odkrycie promieniowania rentgenowskiego w 1895 roku było odkryciem innowacyjnej technologii, otwierającej olbrzymie możliwości w zakresie praktyki medycznej i inicjującej rozwój radiologii i diagnostyki obrazowej. Jednak posiadana wówczas wiedza nie pozwalała przewidzieć nawet najbardziej oczywistych, w świetle współczesnej wiedzy, konsekwencji stosowania długotrwałego, nawet 30 minutowego, naświetlania organizmu49. Nie mamy zatem podstaw do tego, aby oceniać jakie niemiłe niespodzianki mogą nas spotkać w kontekście stosowania innowacyjnych technologii ATMP. Tym bardziej zatem należy wykazać się dużą dozą ostrożności w ocenie ich bezpieczeństwa klinicznego.
Rozpoznanie innowacyjności technologii niesie zatem ze sobą nie tylko zmianę sposobu myślenia o możliwych do podjęcia działaniach medycznych, ale również pewną dozę niepewności co do ich konsekwencji. Jednakże zmiana sposobu myślenia o działaniach medycznych, która dokonała się za sprawą innowacyjnych technologii inżynierii genowej, nie zwalnia od realizacji głównego celu praktyki medycznej, jakim jest zapewnienie bezpieczeństwa pacjenta. Posługujemy się terminem innowacyjne technologie w celu podkreślenia zasadności akceptacji postawy ostrożności epistemicznej. Uznajemy, że mogą wystąpić takie zdarzenia doniosłe klinicznie, które stanowią efekt stosowania innowacyjnych technologii i których nie jesteśmy w stanie przewidzieć na podstawie dostępnych nam modeli teoretycznych i dotychczasowych doświadczeń wyniesionych z badań podstawowych i przedklinicznych. W takiej sytuacji zasadne byłoby, aby niedobór wiedzy o możliwych rezultatach stosowania tych technologii został uzupełniony odpowiednio przeprowadzonymi badaniami klinicznymi. Jest to w istocie klasyczny model rozwoju środka leczniczego, obejmujący trzy główne etapy: badań podstawowych, przedklinicznych i trzech faz badań klinicznych, które zostały zaprezentowane na schemacie poniżej.
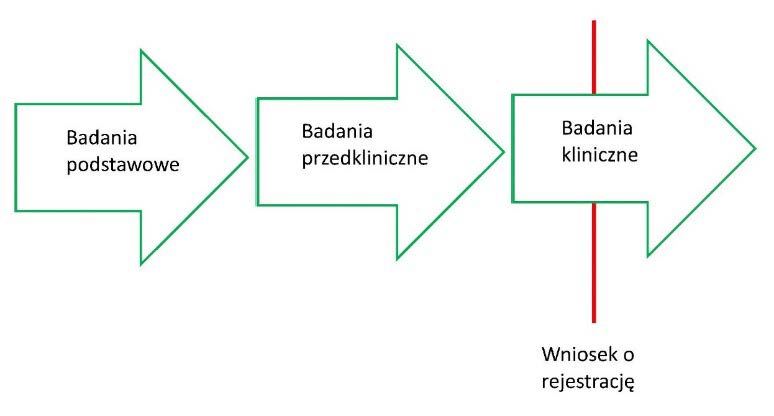
Pomimo różnych zastrzeżeń, problemów i drobnych modyfikacji powyższego schematu, pozostaje on nadal najlepszym z możliwych drogowskazów w procesie oceny bezpieczeństwa i skuteczności leku. Przekonano się bowiem wielokrotnie, że wiedza o mechanizmach działania cząsteczki uzyskiwana w naukach podstawowych może co najwyżej stanowić punkt wyjścia dla stworzenia projektu terapii farmakologicznej poprzez sprecyzowanie mechanizmu i modelu działania środka farmakologicznego oraz opracowanie cząsteczki o określonych właściwościach fizykochemicznych i identyfikację zamierzonych punktów uchwytu dla tej cząsteczki50. Wiedza ta nie wystarcza jednak dla uzyskania satysfakcjonującego stopnia pewności co do efektów klinicznych działania tej cząsteczki. Podobnie wiedzy takiej nie uzyskujemy z modeli zwierzęcych, które, z różnych powodów, dostarczają wyłącznie przybliżenia w ocenie zdarzeń wywołanych zastosowaniem środka farmakologicznego w organizmie ludzkim. W procesach badawczych, których przedmiotem są „klasyczne” środki farmakologiczne, zasadniczy ciężar oceny bezpieczeństwa i skuteczności terapii spoczywa na badaniach klinicznych. W odniesieniu do stosowania technologii innowacyjnych w terapiach ATMP pozyskiwanie danych z badań klinicznych jako danych o kluczowym znaczeniu dla oceny tych terapii wydaje się zatem tym bardziej zasadne. Warto podkreślić, że spełnienie postulatu przeprowadzania badań klinicznych ATMP o najwyższym stopniu rzetelności może okazać się niezwykle trudne51.
Analizując kategorię innowacyjności technologii wykorzystywanych w ATMP nie sposób pominąć kwestii statusu podmiotów wykonujących takie badania. Czy technologie innowacyjne są rozwijane przez podmioty komercyjne (firmy bio-tech), czy niekomercyjne, jak ośrodki badawcze, uniwersytety, instytuty, szpitale? W początkowym okresie, w latach 80-tych i 90 tych XX wieku, rozwój ATMP następował w efekcie intensywnych prac badawczych prowadzonych głównie w dużych ośrodkach uniwersyteckich. Na tym początkowym etapie zaangażowanie firm komercyjnych, w tym również firm farmaceutycznych, w prace badawcze było znikome. Perspektywa aplikacji ATMP u pacjentów wydawała się bowiem niezwykle odległa zarówno z uwagi na ograniczenia technologiczne, jak i brak regulacji legislacyjnych dopuszczających stosowanie ATMP w warunkach praktyki medycznej52.
Zastosowanie nowych biotechnologii dla potrzeb rozwoju ATMP wymagało wykorzystania odpowiedniej infrastruktury technologicznej i odpowiednich kompetencji i wiedzy personelu53. Uniwersytety i placówki badawcze w największym stopniu dysponowały obydwoma tymi elementami54. Fakt ten przekładał się również na rozwój badań klinicznych tych terapii. Do 2015 roku znaczna część badań klinicznych w obszarze ATMP prowadzona była przez podmioty niekomercyjne (73%). Wśród nich przeważał udział uniwersytetów (37% spośród niekomercyjnych) i szpitali (31%). Przewaga podmiotów niekomercyjnych wyraźnie zaznaczała się w I i II fazie badań klinicznych, podczas gdy w fazie III udział jednostek komercyjnych i niekomercyjnych był wyrównany, co ilustruje poniższa tabela55.
Tabela 156
| Podmiot badawczy | Faza I i I/II | Faza II i II/III | Faza III |
| Komercyjny (bio-tech) | 124 (20,5%) | 85 (32,6%) | 35 (53,8%) |
| Niekomercyjny (uniwersytety, instytuty, szpitale) |
480 (79,5%) | 174 (66,6%) | 30 (46,2%) |
| Razem | 604 | 259 | 65 |
Zastosowanie ATMP w początkowym okresie rozwoju biotechnologii było wyraźnie sprofilowane do konkretnego pacjenta. Laboratoria prowadząc badania naukowe pobierały materiał biologiczny (od niego samego lub z innego źródła), modyfikowały go w odpowiedni sposób i aplikowały u chorego. Przetwarzanie produktu odbywało się często z wykorzystaniem unikalnych laboratoryjnych technologii, gwarantujących wysoką, jak na ówczesne standardy, precyzję, ale niewielką wydajność produkcji. Początkowy rozwój ATMP w większym stopniu podporządkowany był raczej realizacji celów badawczych aniżeli powszechnemu wykorzystaniu owych terapii w opiece zdrowotnej57.
Wprowadzenie ATMP dla liczniejszej populacji wymagało opracowania technologii modyfikacji materiału biologicznego charakteryzujących się większą wydajnością produkcji oraz skupienia się na zachowaniu w produkcie wyłącznie tych cech materiału, które są niezbędne dla uzyskania skuteczności i bezpieczeństwa jego aplikacji w szerokiej populacji pacjentów, przy zachowaniu wytycznych unijnych dotyczących dobrych praktyk wytwarzania produktów leczniczych (Good Manufacturing Practice - GMP)58. Wytyczne te obejmują wszystkie aspekty produkcji, poczynając od instrukcji pobierania materiału, poprzez opis procesów szkolenia personelu, a kończąc na ustaleniach dotyczących zasad bezpieczeństwa personelu przy kontaktach z materiałami biologicznymi. Istotne jest w tej sytuacji wypracowanie standardowych procedur operacyjnych (standard operating procedure, SOP), określających kolejne etapy produkcji59. W przypadku najbardziej innowacyjnych biotechnologii wykorzystywanych w ATMP procedury takie nie istniały z uwagi na brak wcześniejszych doświadczeń. Dlatego też zgodność wytwarzania ATMP ze standardami GMP stanowiła w istocie zasadniczy problem stojący na drodze dalszego rozpowszechniania produktów terapii zaawansowanych.
Wskazany powyżej problem łatwo można sobie uzmysłowić odnosząc się chociażby do ekspansywnego rozwoju nowych technologii inżynierii genetycznej, krótko scharakteryzowanych w paragrafie 5. Poszczególne biotechnologie stanowią podstawę dla realizacji nie tylko zupełnie odmiennych strategii terapii, ale dodatkowo w każdej z nich stosowane mogą być dziesiątki różnych materiałów składowych pochodzenia bakteryjnego (wspomniane warianty elementu Cas), wirusowego (wspomniane różne typy wirusów), lub innego. Dlatego też pomimo czasu, jaki upłynął od pierwszych prób i badań nad ATMP, problemy standaryzacji oceny i skuteczności ATMP nie zostały rozwiązane. Cały czas mamy do czynienia z innowacyjnym charakterem stosowanych w nich technologii, co powoduje, że badania nowych ATMP wymagają zaawansowanego zaplecza technologicznego i nadal są w znacznym stopniu ukierunkowane na realizację celów poznawczych. Wystarczy wspomnieć, że spośród 22 badań klinicznych prowadzonych w 2020 roku, które dotyczyły terapii wykorzystujących technologię CRISPR/Cas, aż 16 było przedmiotem badań prowadzonych przez uniwersytety lub szpitale, a tylko wobec pozostałych 6 podejmowano badania prowadzone przez komercyjne ośrodki badawcze60.
Produkty terapii zaawansowanych należą do najbardziej innowacyjnych technik terapii i prewencji we współczesnej medycynie. Nieustannie opracowywane są nowe technologie ATMP. Ważne jest, aby specyfika rozwijanych biotechnologii wykorzystywanych w rozwoju ATMP była uwzględniana w nowych, odpowiednio formułowanych wytycznych dotyczących nadzoru jakości produktów, ich przechowywania, transportu i podawania pacjentowi, w tym również nadzoru biologicznego. Innowacyjność rozwijanych nowych biotechnologii ATMP powoduje, że odpowiedzialność za efekty stosowanych terapii spoczywa przede wszystkim na ośrodkach badawczych, w których opracowuje się nowe ATMP i w których dokonuje się pierwszych aplikacji w badaniach klinicznych. Należy mieć świadomość, że to właśnie ośrodki badawcze przeprowadzające badania podstawowe dysponują kadrą naukową, która posiada największe kompetencje w ocenie i zarządzaniu ryzykiem związanym z rozwojem nowych ATMP. Innowacyjność wymusza zatem nieustanną aktualizację standardów oceny bezpieczeństwa, jednak pamiętać należy, że wytyczne formułowane przez komisje nadzorujące (EMA, CHMP, CAT) są zawsze pochodnymi wobec ustaleń personelu medycznego posiadającego największą wiedzę i doświadczenie w stosowaniu ATMP.
Podkreślić należy, że duża heterogeniczność ATMP skutkuje tym, że nie można opracować ogólnych wytycznych i procedur związanych z ich wytwarzaniem, przetwarzaniem, przechowywaniem i stosowaniem. Jest to zupełnie nowe wyzwanie, które wynika bezpośrednio z innowacyjności i szybkiego rozwoju ATMP. Nie mamy do czynienia z klasą produktów, dla których można określić regulacje, które nie będą podlegały zmianom z uwagi na niezmienność stosowanych metod wytwarzania i wprowadzania produktów na rynek. W konsekwencji trudne jest również modelowanie ryzyka dla produktów ATMP, w szczególności niejednoznaczna jest ocena ilorazu korzyść / ryzyko z uwagi na duże zróżnicowanie czynników ryzyka, które nie występują w przypadku produktów klasycznej farmakologii z udziałem cząsteczek biologicznie aktywnych61.
ATMP cały czas należą do najbardziej nowatorskich terapii medycznych. Dlatego też ciągle mamy do czynienia z pewnym nienadążaniem regulacji legislacyjnych za wprowadzanymi nowymi technologiami. Problemy te spotęgowane są dodatkowo faktem, że terapie ATMP mają nadal charakter zindywidualizowanych terapii62. Wytwarzanie produktów terapii zaawansowanych często nie ma zatem charakteru przemysłowego, lecz obejmuje małe ich ilości. Produkcja odbywa się w ośrodkach badawczych, które mogą posługiwać się różnymi procedurami postępowania. Próby stworzenia regulacji prawnych muszą zagwarantować bezpieczeństwo pacjentów, nie umniejszając jednak innowacyjności wprowadzanych nowych technologii ATMP63. Zachowana musi zatem być pewna elastyczność formułowanych regulacji. Aby zrealizować te cele konieczne jest stworzenie platform umożliwiających wymianę informacji pomiędzy (1) ośrodkami naukowymi, w których prowadzone są badania nad nowymi technikami ATMP, (2) przemysłem farmaceutycznym i sponsorami badań oraz (3) agencjami regulacyjnymi: CAT, CHMP, EMA, FDA. Translacja kliniczna nowych ATMP musi być postrzegana jako efekt współpracy wszystkich stron zaangażowanych w ich tworzenie i stosowanie64. Konieczne jest umożliwienie przepływu informacji pomiędzy interesariuszami prezentującymi różne perspektywy badań, zarówno podmiotami komercyjnymi, jak i niekomercyjnymi. Musi zatem nastąpić pewne zrównoważenie ustaleń legislacyjnych. Rozporządzenia nie mogą hamować rozwoju nowych technologii, ale z drugiej strony ich stopniowe wprowadzanie jest konieczne dla zapewnienie bezpieczeństwa i ograniczenia nadużyć związanych z chęcią osiągnięcia korzyści finansowych. Celem jest wymiana informacji pomiędzy instytucjami badawczymi (takimi jak uniwersytety czy instytuty) a podmiotami komercyjnymi. Te pierwsze dysponują najbardziej rozwiniętą wiedzą odnośnie do nowatorskich technologii stosowanych w procesie wytwarzania ATMP, te drugie z kolei posiadają wiedzę typu „know-how”, dotyczącą procesów logistycznych: dystrybucji, przechowywania i wdrażania procedur aplikacji produktów ATMP.
Goula et al. (2020): 781. ↑
Rzepiński (2023a). ↑
Goula et al. (2020): 781. ↑
European Commission (2009), cz. IV, pkt. 2.1; Zobel, Heelan (2017); Hanna et al. (2016): 3; Detela, Lodge (2019): 208. ↑
Hildt (2016). ↑
European Commission (2009), cz. IV, pkt. 2.2. ↑
Zobel, Heelan (2017): 1; Detela, Lodge (2019): 223; Buckland, Gaspar (2014). ↑
Rozporządzenie 1394 (2007): Rozdział 1. ↑
Rozporządzenie 1394 (2007): Rozdział 1; Hanna et al. (2016): 3; Sanzenbacher et al. (2007); European Commission (2009). ↑
Rozporządzenie 1394 (2007): Rozdział 1. ↑
Gonçalves (2020): 312. ↑
Sagar et al. (2019). ↑
Detela, Lodge (2019): 209. ↑
European Commission (2003). ↑
Rozporządzenie 1394 (2007): Rozdział 1; Gonçalves (2020): 311. ↑
Rozporządzenie 1394 (2007). ↑
Goula et al. (2020): 783. ↑
Carvalho, Sepodes, Martins (2017): 2. ↑
Rozporządzenie 1394 (2007): pkt. 13, 14. ↑
Ibidem: pkt. 16, 20. ↑
Blaese et al. (1995); Bordignon et al. (1995). ↑
Raper et al. (2003). ↑
Cavazzana-Calvo, Fisher (2000). ↑
Detela, Lodge (2019): 224. ↑
Pellegrini et al. (2018); Hanna et al. (2016): 2. ↑
Detela, Lodge (2019): 218. ↑
Hanna et al. (2016): 3. ↑
Schemat za: Hanna et al. (2016). ↑
Schemat za: Hanna et al. (2016). ↑
Fry et al. (2018); Susanibar-Adaniya, Cohen, Garfall (2019); Raje et al. (2019). ↑
Gromadzka et al. (2010). ↑
Uddin, Rudin, Sen (2020): 2. ↑
Cong et al. (2013). ↑
EMA (2014); Detela, Lodge (2019): 211. ↑
Rosenberg et al. (1990). ↑
Fire et al. (1998). ↑
Mojica et al. (2000). ↑
Cong et al. (2013). ↑
Clin.Trial.gov. (2018). ↑
Uddin, Rudin, Sen (2020): 3. ↑
Li, Mei, Hu (2020): 177–178; Maeder et al. (2019). ↑
Poza wirusami funkcje wektorów mogą pełnić również plazmidy bakteryjne, liposomy, mRNA. ↑
Uddin, Rudin, Sen (2020): 5. ↑
Trounson, McDonald (2015); Goradel et al. (2017). ↑
Schemat za: Uddin, Rudin, Sen (2020): 3. ↑
Buckland, Gaspar (2014): 163. ↑
Uzyskane doświadczenie można jednak wykorzystać w celu podjęcia odpowiedniego przeciwdziałania, np. dokonując odpowiednich modyfikacji genetycznych w wektorze poprzez wprowadzenie włącznika genu samobójczego, jak w terapii ZalmoxisuR. ↑
Rzepiński (2023b). ↑
Gutt (1995): 337–338. ↑
Rzepiński (2019). ↑
Rzepiński (2023a). ↑
Goula et al. (2020): 784. ↑
Gastelurrutia et al. (2021): 2. ↑
Buckland, Gaspar (2014): 163. ↑
Hanna et al. (2016): 6. ↑
Dane za: Trounson, McDonald (2015). Na tę kwestię zwracają również uwagę Buckland, Gaspar (2014): 164; Maciulaitis et al. (2012). ↑
Goula et al. (2020): 784. ↑
Ibidem: 784; Pizevska et al. (2022): 2. ↑
Gastelurrutia et al. (2021): 3. ↑
Uddin, Rudin, Sen (2020): 11–12. ↑
Pizevska et al. (2022): 4. ↑
Goula et al. (2020): 783. ↑
Pizevska et al. (2022). ↑
Ibidem: 6. ↑
Podziękowania: Autor składa podziękowania Profesorowi Włodzimierzowi Galewiczowi za możliwość wzięcia udziału w konferencji „O granicach eksperymentalnych terapii”.
Finansowanie: Nie dotyczy.
Konflikt interesów: Autor oświadcza, że w ramach pracy nad artykułem nie wystąpił konflikt interesów na żadnym z jej etapów.
Licencja: Artykuł opublikowany w otwartym dostępie na licencji Creative Commons Attribution License, która dopuszcza użycie, rozpowszechnianie oraz powielanie w dowolnym medium, pod warunkiem, że oryginalne dzieło jest stosownie cytowane.
Blaese R., Culver K.W., Miller A.D., Carter C.S., Fleisher T., Clerici M., Shearer G., Chang L., Chiang Y., Tolstoshev P., Greenblatt J.J., Rosenberg S.A., Klein H., Berger M., Mullen C. A., Ramsey W.J., Muul L., Morgan R.A., Anderson W.F. (1995), T lymphocyte-directed gene therapy for ADASCID: Initial trial results after 4 years, „Science” 270: 475–480, URL = https://doi.org/10.1126/science.270.5235.475 [dostęp 07.08.2023].
Bordignon C., Notarangelo L.D., Nobili N., Ferrari G., Casorati G., Panina P., Mazzolari E., Maggioni D., Rossi C., Servida P., Ugazio A.G., Mavilio F. (1995), Gene therapy in peripheral blood lymphocytes and bone marrow for ADA- immunodeficient patients, „Science” 270: 470–475, URL = https://doi.org/10.1126/science.270.5235.470 [dostęp 07.08.2023].
Buckland K., Gaspar H.B. (2014), Gene and cell therapy for children – New medicines, new challenges?, „Advanced Drug Delivery Reviews” 73 (100): 162–169, URL = https:// doi.org/10.1016/j.addr.2014.02.010 [dostęp 07.08.2023].
Carvalho M., Sepodes B., Martins A.P. (2017), Regulatory and Scientific Advancements in Gene Therapy: State-of-the-Art of Clinical Applications and of the Supporting European Regulatory Framework, „Frontiers in Medicine (Lausanne)” 4: 182, URL = https://doi.org/10.3389/fmed.2017.00182 [dostęp 07.08.2023].
Cavazzana-Calvo M., Fisher A. (2000), Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease, „Science” 288: 669–672, URL = https://doi.org/10.1126/science.288.5466.669 [dostęp 07.08.2023].
ClinicalTrials.gov (2018), A Safety and Efficacy Study Evaluating CTX001 in Subjects With Severe Sickle Cell Disease, URL = https://clinicaltrials.gov/ct2/show/NCT03745287 [dostęp 18.06.2023].
Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. (2013), Multiplex genome engineering using CRISPR/Cas systems, „Science” 339: 819–823, URL = https://doi.org/10.1126/science.1231143 [dostęp 07.08.2023].
Detela G., Lodge A. (2019), EU Regulatory Pathways for ATMPs: Standard, Accelerated and Adaptive Pathways to Marketing Authorisation, „Molecular Therapy: Methods & Clinical Development” 13: 205–232, URL = https://doi.org/10.1016/j.omtm.2019.01.010 [dostęp 07.08.2023].
EMA (European Medicines Agency) (2014), Mandate of the EMA Innovation Task Force (ITF), URL = https://www.ema.europa.eu/en/documents/other/mandate-european-medicines-agency-innovation-task-force-itf_en.pdf [dostęp 01.07.2023].
European Commission (2003), EU directives. Commission Directive 2003/63/EC of 25 June 2003 amending Directive 2001/83/EC of the European Parliament and of the Council on the Community code relating to medicinal products for human use, „Official Journal of the European Union” L159: 46–94, URL = https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=uriserv%3AOJ.L_.2003.159.01.0046.01.ENG&toc=OJ%3AL%3A2003%3A159%3ATOC [dostęp 07.08.2023].
European Commission (2009), Commission Directive 2009/ 120/EC of 14 September 2009 amending Directive 2001/83/ EC of the European Parliament and of the Council on the Community code relating to medicinal products for human use as regards advanced therapy medicinal products, „Official Journal of the European Union” L242: 3–12, URL = https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=uriserv%3AOJ.L_.2009.242.01.0003.01.ENG&toc=OJ%3AL%3A2009%3A242%3ATOC [dostęp 07.08.2023].
Fire A., Xu S., Montgomery M.K., Kostas S.A., Driver S.E., Mello C.C. (1998), Potent and specific genetic interference by double- stranded RNA in Caenorhabditis elegans, „Nature” 391: 806–811, URL = https://doi.org/10.1038/35888 [dostęp 07.08.2023].
Fry T., Shah N.N., Orentas R.J. et al. (2018), CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19- targeted CAR immunotherapy, „Nature Medicine” 24: 20–28, URL = https://doi.org/10.1038/nm.4441 [dostęp 07.08.2023].
Gastelurrutia P., Prat-Vidal C., Vives J., Coll R., Bayes-Genis A., Galvez-Monton C. (2021), Transitioning From Preclinical Evidence to Advanced Therapy Medicinal Product: A Spanish Experience, „Frontiers in Cardiovascular Medicine” 8: 1-10, URL = https://doi.org/10.3389/fcvm.2021.604434 [dostęp 07.08.2023].
Gonçalves E. (2020), Advanced therapy medicinal products: value judgement and ethical evaluation in health technology assessment, „The European Journal of Health Economics” 21 (3): 311–320, URL = https://doi.org/10.1007/s10198-019-01147-x [dostęp 07.08.2023].
Goradel N.H., Ghiyami-Hour F., Negahdari B., Malekshahi Z.V., Hashemzehi M., Masoudifar A., Mirzaei H. (2017), Stem Cell Therapy: A New Therapeutic Option for Cardiovascular Diseases, „Journal of Cellular Biochemistry” 119 (1): 95–104, URL = https://doi.org/10.1002/jcb.26169 [dostęp 07.08.2023].
Goula A., Gkioka V., Michalopoulos E., Katsimpoulas M., Noutsias M., Sarri F., Stavropoulos C., Kostakis A. (2020), Advanced Therapy Medicinal Products Challenges and Perspectives in Regenerative Medicine, „Journal of Clinical Medial Research” 12 (12): 780–786, URL = https://doi.org/10.14740/jocmr3964 [dostęp 07.08.2023].
Gromadzka A., Kubiczak M., Jankowska A. (2010), Terapia genowa i jej zastosowanie w leczeniu nowotworów ginekologicznych, „Ginekologia Polska” 81 (1): 50–54.
Gutt R. (1995), Rozwój klinicznej medycyny wewnętrznej i specjalności pokrewnych, [w:] Historia medycyny, T. Brzeziński (red.), PZWL, Warszawa.
Hanna E., Rémuzat C., Auquier P., Toumi M. (2016), Advanced therapy medicinal products: current and future perspectives, „Journal of Market Access & Health Policy” 4 (1): 1-10, URL = https://doi.org/10.3402/jmahp.v4.31036 [dostęp 07.08.2023].
Hildt E. (2016), Human Germline Interventions – Think First, „Frontiers in Genetics” 7 (81): 1–3, URL = https://doi.org/10.3389/fgene.2016.00081 [dostęp 07.08.2023].
Li C., Mei H., Hu Y. (2020), CAR-T Applications and explorations of CRISPR/Cas9 in CAR T-cell therapy, „Briefings in Functional Genomics” 19 (3): 175–182, URL = https://doi.org/10.1093/bfgp/elz042 [dostęp 07.08.2023].
Maciulaitis R., D’Apote L., Buchanan A., Pioppo L., Schneider C.K. (2012), Clinical development of advanced therapy medicinal products in Europe: Evidence that regulators must be proactive, „Molecular Therapy” 20 (3): 479–482, URL = https://doi.org/10.1038/mt.2012.13 [dostęp 07.08.2023].
Maeder M., Stefanidakis M., Wilson C.J. et. al. (2019), Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10, „Nature Medicine” 25: 229–233, URL = https://doi.org/10.1038/s41591-018-0327-9 [dostęp 07.08.2023].
Mojica F., Díez-Villaseñor C., Soria E., Juez G. (2000), Biological significance of a family of regularly spaced repeats in the genomes of archaea, bacteria and mitochondria, „Molecular Microbiology” 36 (1): 244–246, URL = https://doi.org/10.1046/j.1365-2958.2000.01838.x [dostęp 07.08.2023].
Pellegrini G., Ardigò D., Milazzo G., Iotti G., Guatelli P., Pelosi D., De Luca M. (2018), Navigating Market Authorization: The Path Holoclar Took to Become the First Stem Cell Product Approved in the European Union, „Stem Cells Translational Medicine” 7 (1): 146–154, URL = https://doi.org/10.1002/sctm.17-0003 [dostęp 07.08.2023].
Pizevska M., Kaeda J., Fritsche E., Elazaly H., Reinke P., Amini L. (2022), Advanced Therapy Medicinal Products’ Translation in Europe: A Developers’ Perspective, „Frontiers in Medicine” 9: 1-8, URL = https://doi.org/10.3389/fmed.2022.757647 [dostęp 07.08.2023].
Raje N., Berdeja J., Lin Y. et al. (2019), Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma, „New England Journal of Medicine” 380 (18): 1726–1737, URL = https://doi.org/10.1056/NEJMoa1817226 [dostęp 07.08.2023].
Raper S., Chirmule N., Lee F.S., Wivel N.A., Bagg A., Gao G.., Wilson J.M., Batshaw M.L. (2003), Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer, „Molecular Genetics and Metabolism” 80 (1–2): 148–158, URL = https://doi.org/10.1016/j.ymgme.2003.08.016 [dostęp 07.08.2023].
Rosenberg S., Aebersol P., Cornetta K., Kasid A., Morgan R., Moen R., Karson E., Lotze M., Yang J., Topalian S., Merino M., Culver K., Miller D., Blaese M., Anderson F., (1990), Gene transfer into humans–immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction, „New England Journal of Medicine” 323: 570–578, URL = https://doi.org/10.1056/nejm199008303230904 [dostęp 07.08.2023].
Rozporządzenie 1394 (2007) – Regulation (EC) No 1394/2007 of the European Parliament and of the council of 13 November 2007 on advanced therapy medicinal products and amending Directive 2001/83/EC, URL = https://eurlex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2007:324:0121:0137:en:PDF [dostęp 21.01.2023].
Rzepiński T. (2019), Odkrycie środka leczniczego – dwie koncepcje rozwoju badań w zakresie farmakologii przedklinicznej, [w:] Genius Vitae. Księga pamiątkowa dedykowana panu profesorowi Marianowi Józefowi Wnukowi, S. Janeczek, Z Wróblewski, A. Starościc, (red.) Wydawnictwo KUL, Lublin: 283–301.
Rzepiński T. (2023a), Problemy i ograniczenia badań klinicznych produktów leczniczych terapii zaawansowanych (ATMP), Ekspertyza dla Stałej Rektorskiej Komisji ds. Etyki Badań Naukowych UJ, URL = https://etykabadan.komisja.uj.edu.pl/dzialalnosc/przeglady-i-ekspertyzy [dostęp 07.08.2023].
Rzepiński T. (2023b), Subjectivity of pre-test probability value: controversies over the use of Bayes’ Theorem in medical diagnosis, „Theoretical Medicine and Bioethics” 44: 301–324 (w druku), URL = https://doi.org/10.1007/s11017-023-09614-6 [dostęp 07.08.2023].
Sagar R., Götherström C., David A.L., Westgren M. (2019), Fetal stem cell transplantation and gene therapy, „Best Practice & Research Clinical Obstetrics and Gynaecology” 58: 142–153, URL = https://doi.org/10.1016/j.bpobgyn.2019.02.007 [dostęp 07.08.2023].
Sanzenbacher R., Dwenger A., Schuessler-Lenz M., Cichutek K., Flory E. (2007), European regulation tackles tissue-engineering, „Nature Biotechnology” 25 (10): 1089–1091, URL = https://doi.org/10.1038/nbt1007-1089 [dostęp 07.08.2023].
Susanibar-Adaniya S.P., Cohen A., Garfall A.L. (2019), Chimeric antigen receptor T cell immunotherapy for multiple myeloma: a review of current data and potential clinical applications, „American Journal of Hematology” 94 (1): 28–33, URL = https://doi.org/10.1002/ajh.25428 [dostęp 07.08.2023].
Trounson A., McDonald C. (2015), Stem Cell Therapies in Clinical Trials: Progress and Challenges, „Cell Stem Cell” 17 (1): 11–22, URL = https://doi.org/10.1016/j.stem.2015.06.007 [dostęp 07.08.2023].
Uddin F., Rudin C.M., Sen T. (2020), CRISPR Gene Therapy: Applications, Limitations, and Implications for the Future Fathema, „Frontiers in Oncology” 10: 1-17, URL = https://doi.org/10.3389/fonc.2020.01387 [dostęp 07.08.2023].
Zobel A., Heelan B. (2017), Regulatory, clinical and logistics challenges of ATMPs in clinical research, „European Pharmaceutical Review” 4, URL = https://www.europeanpharmaceuticalreview.com/article/81122/regulatory-clinical-and-logistics-challenges-of-advanced-therapy-medicinal-products-atmps-in-clinical-research/ [dostęp 08.07.2023].